|
Picard HsMetrics
|
Enables Picard HsMetrics generation. When enabled, select the probe BED type. If Use Custom Probe BED is chosen, select the custom probe BED file to use for the analysis.
|
|
Somatic Variant Calling
|
Enables additional parameter settings when somatic small variant calling is enabled.
|
•
|
Liquid Tumor—Enable liquid tumor mode for variant calling. This mode adjusts the small variant caller to allow variants to be called in the presence of tumor-in-normal contamination.
|
|
•
|
Allele Frequency Filtering—Enable allele frequency filtering for somatic variant calling. |
|
•
|
Somatic variant frequency call threshold (percentage)—Somatic variants with an allele frequency less than this threshold level are not called. Low threshold values might result in more false positive variants. The range of this setting is 0–30. The default value is 1.
Decreasing this value increases variant caller sensitivity but raises the risk of false positives. |
|
•
|
Somatic variant frequency filter threshold (percentage)—Somatic variants with an allele frequency less than this threshold level are marked as filtered. The range of this setting is 0–30. The default value is 5. |
|
•
|
Somatic Quality Filtering—Enable filtering by somatic quality score (SQ). |
|
•
|
Somatic variant quality call threshold—Somatic variants with a quality score less than this threshold level are not called. Low threshold values might result in more false positive variants. The range of this setting is 0–30. The default value is 1. |
|
•
|
Somatic variant quality filter threshold—Somatic variants with a quality score less than this threshold level are marked as filtered. The range of this setting is 0–30. The default value is 3. |
|
•
|
Baseline Systematic Noise BED—Select a baseline systematic noise BED file for analysis. Systematic Noise Detection is automatically enabled when a BED file is specified. This app supports only BED files created by the DRAGEN Baseline Builder app. |
|
•
|
Somatic Hotspots—Specify whether to use built-in regions matched to the selected reference or a VCF input with custom hotspot regions. |
|
•
|
[Optional] Excluded Regions—Ignore small variants that occur in certain regions. Select a BED file that defines the regions you want to exclude. |
|
•
|
Tumor Mutational Burden—Use one QC Coverage Region and enable Illumina Annotation Engine annotation. Cannot be enabled when a custom reference genome is used. The TMB calculation might not be reliable for small panels (< 1 Megabase in size).
[Optional] Select a BED file (in *.bed or .bed.gz format) that defines the genomic regions to calculate TMB in. If not supplied, TMB is calculated using the selected enrichment regions.
|
|
•
|
Germline Tagging—Use population databases to tag possible germline variants with GermlineStatus in the INFO field. Current databases include 1KG, both exome and genome sequencing data from gnomAD. Illumina Annotation Engine annotation is automatically turned on when this setting is enabled. |
|
•
|
Multi-allelic Filtering—Enable the multiallelic filter for Somatic mode. Enabled by default. |
|
•
|
QC Somatic Contamination VCF—Use a VCF with population allele frequencies to calculate the fraction of reads that come from cross-sample contamination. Applicable only when using a human reference built into the appication.
|
|
|
CNV
|
Enables CNV analysis. If enabled, configure the following settings.
|
•
|
Reference Calls—Include copy neutral (REF) calls in the output CNV VCF. |
|
•
|
Heterogeneous Shifting Levels Model—A variant of the shifting level model. Recommended for use in exome analysis and other capture kits that are not equally spaced. |
|
•
|
Circular Binary Segmentation—Iteratively identifies change points in a genomic sequence using a nonparametric hypothesis testing approach. |
|
•
|
Shifting Levels Model—Models genomic data as the sum of two independent stochastic processes and segments using a subclass of Hidden Markov Model. |
|
•
|
Targeted Segmentation (Segment BED)—In applications for targeted panels, you can limit the segmentation and calling performed on intervals by specifying a CNV segmentation BED file. |
|
•
|
CNV caller quality filter threshold—Specifies the QUAL value at which variants are filtered in the CNV VCF. The range of this setting is 0–90.0. The default value is 50.0. |
|
•
|
CNV caller copy ratio filter threshold—Specifies the minimum value, centered about 1.0 at which a reported event is marked as PASS in the output VCF file. The range of this setting is 0.0–1.0. The default value is 0.2. This default value leads to calls with a CR ≤ 8 or a CR ≥ 1.2. |
|
•
|
CNV Baseline Files—Select the CNV baseline files. You can select up to 99 files. All files must be of the same type (eg, all *.target.counts or all *.target.counts.gc-corrected). You can create CNV baselines using the DRAGEN CNV Baseline Builder app. |
|
•
|
[Optional] Custom CNV BED File—Select a BED file that indicates the target intervals to use for sample coverage. If this file is not specified, default enrichment regions are used. |
|
•
|
GC Bias Correction—Enabled by default. |
|
|
HLA Typing
|
Enables human leukocyte antigen (HLA) alignment and HLA-specific analysis components for class I HLA allele typing. The DRAGEN HLA caller enables HLA genotyping at two-field resolution (also known as four-digit typing).
|
|
Structural Variants
|
Enables structural variant analysis.
|
•
|
DNA Fusion Filter—Enable DNA Fusion Filter Calling, which might reduce false positives. |
|
•
|
Forced Genotyping SV VCF—Select an SV VCF (in *.vcf.gz format) for forced genotyping of SV alleles. Variants from this VCF file are scored and output from the SV caller even if the variant is not supported in the sample data. Forced genotyping typically enables known variants to be detected at higher recall than standard variant discovery (particularly for variant discovery on a lower-depth sample). |
|
•
|
ITD Region BED—Select a BED file (in *.bed or *.bed.gz format) for targeted calling of internal tandem duplicates (ITD). This option replaces the standard SV BED setting. SV calls are not output elsewhere in the genome. SV caller settings are more sensitive within this region. |
|
•
|
Locus Node Target Region BED—Select a BED file (in *.bed or *.bed.gz format) for targeted calling of locus node target regions. This replaces the standard SV BED setting so SV calls will output elsewhere in the genome. SV caller settings are modified for increased sensitivity within this region. |
|
|
UMI Settings
|
Enables UMI-based read processing when the run is configured for the Map/Align pipeline configuration. Disabled by default. If enabled, set the following options:
|
•
|
UMI Library Type—Specify the type of UMI used for the data (nonrandom-duplex, random-duplex, or random-simplex). |
|
•
|
UMI-Aware Variant Calling—Specify the type of variant calling to run on UMI data, if any. These presets override other somatic variant calling settings such as filtering thresholds. |
|
•
|
High depth—Enables somatic variant calling with presets optimized for liquid biopsy analysis with post-collapsed coverage rates of roughly 2000-2500X and target allele frequencies of 0.4% and higher (--vc-enable-umi-liquid). |
|
•
|
Low Depth—Enables somatic variant calling with presets optimized for solid tumor analysis with post-collapsed coverage rates of roughly 200-300X and target allele frequencies of 5% and higher (--vc-enable-umi-solid). |
|
•
|
Germline—Enables variant calling for germline UMI data (--vc-enable-umi-germline). |
|
•
|
UMI Min Supporting Reads—Set the minimum number of supporting reads required for a family. Default value is 2. |
|
•
|
UMI Error Correction Table—Select the lookup table for UMI correction. If a lookup table is not specified, the Illumina nonrandom UMIs from the TruSight Oncology 170 RUO, TruSight Oncology 500 RUO, and IDT for Illumina - UMI DNA Index Anchors kits are used. |
|
•
|
UMI Nonrandom Whitelist—Select the file containing a list of valid, nonrandom UMIs. Each UMI in the file must be on separate lines. This setting cannot be used if a UMI error correction table was selected. |
|
|
Advanced Settings
|
|
•
|
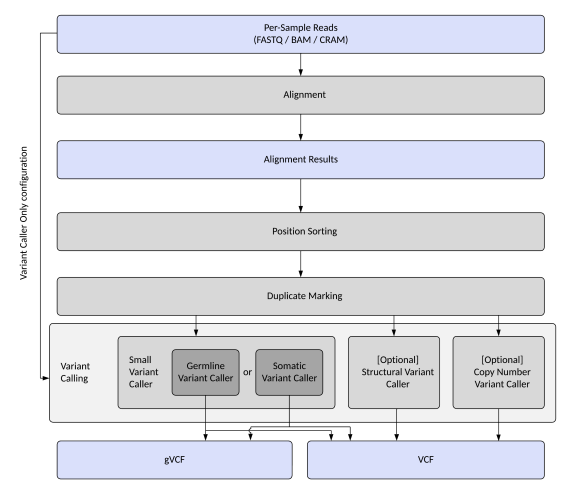
Map/Align + Variant Caller—Samples are mapped, aligned to the reference genome, and position-sorted. Variant calling is also performed. |
|
•
|
Variant Caller Only—Only variant calling is performed. This configuration only accepts BAM input files. |
|
•
|
Small Variant Caller Output
|
|
•
|
VCF and GVCF—Variants are recorded individually and nonvariants are grouped into blocks. |
|
•
|
VCF and GVCF with BP_RESOLUTION—Variants and nonvariants are recorded individually. This option is typically used for debugging and increases run time and gVCF file size. |
When the small variant is set to Somatic mode, the output format is VCF.
|
•
|
Base Padding (for Variant Calling)—Enter a nonnegative integer to define the base padding to add to each target BED region. This option is used to pad targeted regions for variant calling and does not affect most enrichment metrics. |
|
•
|
Duplicate Marking—Enabled by default. Disabled when UMI read processing is enabled. |
|
•
|
Nirvana Annotation—Output a JSON file with annotations for variants in all output DRAGEN VCFs. Disabled by default. |
|
•
|
MAF Output—When Nirvana Annotation is enabled, you can also output annotations in Mutation Annotation Format (MAF). |
|
•
|
ForceGT VCF—Select a *.vcf or *.vcf.gz file of small variants to force genotype. |
|
•
|
Variant Calls Hard Filter—Enter an expression to apply custom filtering to the hard filtered VCF output. The default expression is: DRAGENHardQUAL:all:QUAL<10.4139;LowDepth:all:DP<1
|
|
•
|
High Coverage Mode—Prevent the small variant caller from subsampling reads. This enables the small variant caller to consider up to 100,000 reads per locus. Enabling this mode increases run time. |
|
•
|
Low Pass Targeting Sequencing—Remove the default variant calling flags that are better suited for higher depth targeted sequencing. Enabling this setting might provide better results for low-depth sequencing applications. |
|
•
|
Combine Phased Variants—Enter a base pair distance over which phased variants are combined. Default value is 15. |
|
•
|
QC Coverage Calculation—When selected, alignments are resolved to fragments and are not double-counted. Selecting this option increases run time. |
|
•
|
Bin Memory—Specify the total memory (in GB) used for partitioning sort data. The range of this setting is 20–200. The default value is 40. |
|
•
|
CRAM Decompression Reference—Select a reference genome to realign a CRAM input that was created with a different reference. |
|
•
|
CRAM Custom Reference—Specify a custom FASTA file to use as the CRAM reference. The FASTA index file (*.fai) must be present with the FASTA file. |
|
|
Beta Features
|
The settings in this section are in beta; use this field with caution. Enabling any of these features may cause the analysis to abort.
|
•
|
Fragmentomics—When enabled, the application outputs fragment profile metrics. |
|
•
|
Fragmentomics Window Protection Score—Specify a BED file that defines the regions in which to use to calculate a Window Protection Score. The file must have the chr, start, and end columns only. |
|
•
|
Fragmentomics Exclude Regions—Specify a BED file that defines the regions to exclude from Fragmentomics analysis. The file must have the chr, start, and end columns only. |
|
•
|
Enrichment High Sensitivity SNV Analysis—The following DRAGEN settings for Enrichment high sensitivity UMI enabled assay for SNV are enabled |
|
•
|
Upload Baseline Systematic Noise Bed file |
All other SNV settings are disabled.
|
•
|
Enrichment High Sensitivity CNV Analysis—DRAGEN settings for Enrichment high sensitivity UMI enabled assay for CNV are enabled |
|
•
|
Enrichment High Sensitivity SV Analysis—DRAGEN settings for Enrichment high sensitivity UMI enabled assay for SV (Fusions) are enabled |
|
|
Batching Options
|
When enabled, the application queues multiple inputs per node and processes them sequentially. This option is available only when using biosample FASTQ input files.
|
|
Additional Arguments
|
For advanced users only; use this field with caution. Enter DRAGEN command-line arguments for additional analysis information.
|
|
Automation Settings
|
Enables automation settings. Specify a sample by selecting the option that matches the input file type and the sex. These settings should only be used when launching an app using a biosample workflow or BaseSpace Sequence Hub CLI.
|