Parsing Sequencing Meta-Data into Clarity LIMS
Once a sequencing run has occurred, there is often a requirement to store the locations of the FASTQ / BAM files in BaseSpace Clarity LIMS.
For paired-end sequencing, it is likely that the meta-data file that describes the locations of these files will contain two rows for each sample sequenced: one for the first read, and another for the second read.
Such a file is illustrated here:

Column 2 of the file, Sample ID, contains the LIMS IDs of the artifacts for which we want to store the FASTQ file values listed in column 3 (Fastq File).
This example discusses the strategy for parsing and storing data against process inputs, when that data is represented by multiple lines in a data file.
The attached script will parse a data file containing multiple lines of FASTQ file values, and will store the locations of those FASTQ files in user-defined fields.
Process Configuration
In this example, the process is configured to have a single shared ResultFile output.
Parameters
The EPP command is configured to pass the following parameters:
|
-u |
The username of the current user (Required) |
The {username} token |
|
-p |
The password of the current user (Required) |
The {password} token |
|
-s |
The URI of the step that launches the script (Required) |
The {stepURI:v2:http} token |
|
-i |
The limsid of the shared ResultFile output (Required) |
The {compoundOutputFileLuid0} token |
An example of the full syntax to invoke the script is as follows:
/usr/bin/python /opt/gls/clarity/customextensions/parseMetadata.py -l 24-9953 -u admin -p securepassword -s http://192.168.9.123:8080/api/v2/steps/24-9953 -i 92-20553
The user-interaction is comprised of the following steps:
Step 1
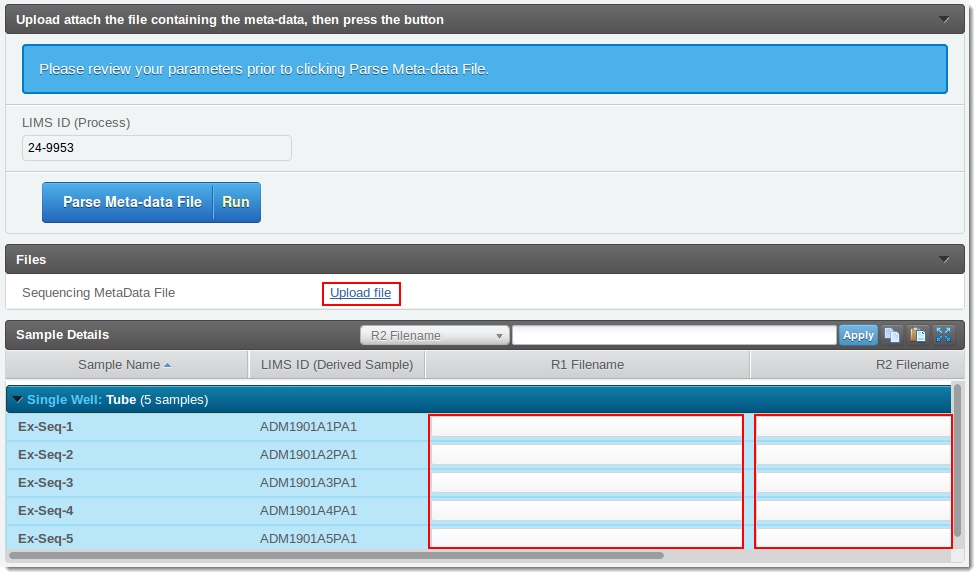
The user runs the process up to the Record Details screen as shown in the following image. Note that initially:
| • | The Sequencing meta-data file is still to be uploaded. |
| • | The values for the R1Filename and R2 Filename fields are empty. |
Step 2
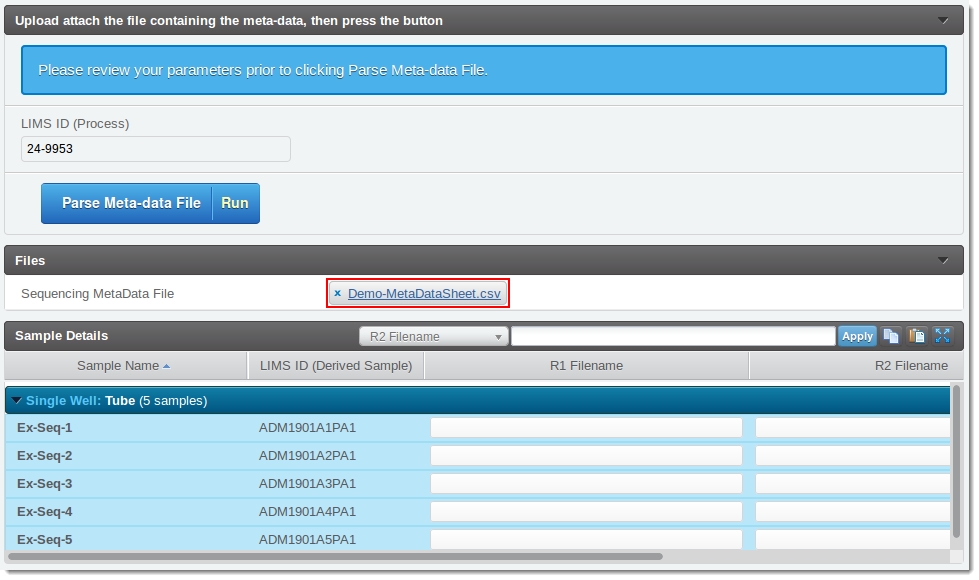
The user clicks Upload file and attaches the meta-data file. Once attached, the user's screen will resemble this:
Step 3
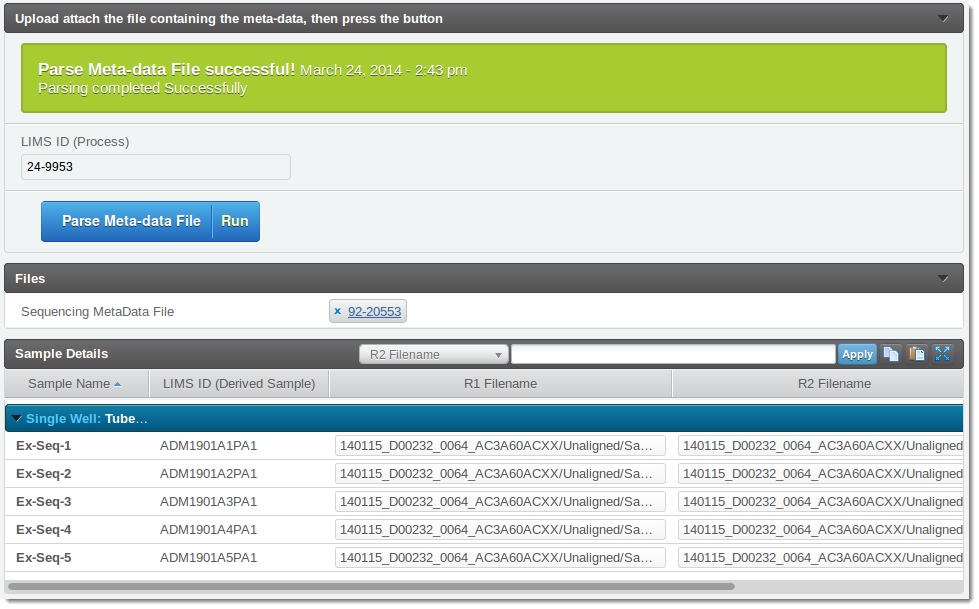
Now that the meta-data file is attached, the user clicks Parse Meta-data File. This invokes the parsing script.
If parsing was successful, the user's screen will resemble the next image.
Note that the values for the R1Filenames and R2 Filenames have been parsed from the file and will be stored in Clarity LIMS.
The key methods of interest are main(), parseFile() and fetchFile(). The main() method calls parseFile(), which in turn calls fetchFile().
fetchFile() method
The fetchFile() method relies upon the fact that the script is running on the Clarity LIMS server, and as such has access to the local file system in which the file (uploaded in Step 2) now resides.
Thus, fetchFile() can use the API to:
| 1. | Convert the LIMSID of the file to the location on disk. |
| 2. | Copy the file to the local working directory, ready to be parsed by parseFile(). |
parseFile() method
| 1. | The parseFile() method creates two data structures that are used in the subsequent code within the script: |
| • | The COLS dictionary has the column names from the first line of the file as its key, and the index of the column as the value. |
| • | The DATA array contains each subsequent line of the file as a single element. Note that this parsing logic is overly-simplistic, and would need to be supplemented in a production environment. For example, if the CSV file being parsed does not have the column names in the first row, then exceptions would likely occur. Similarly, we assume the file being parsed is CSV, and likewise any data elements which themselves contain commas would likely cause a problem. For the sake of clarity such exception handling has been omitted from the script. |
| 2. | Once parseFile() has executed successfully the inputs to the process that has invoked the script are gathered, and 'batch' functionality is used to gather all of the artifacts in a single batch-retrieve transaction. |
| 3. | All that remains is to step through the elements within the DATA array, and for each line, gather the values of the Fastq File and Sample ID columns. For each Sample ID value: |
| • | The corresponding artifact is retrieved. |
| • | Depending on whether the value of the Fastq File column represents the filename for the first or second read, either the R1 Filename or the R2 Filename user-defined field is updated. |
| 4. | Once the modified artifacts have been saved, the values will display in the Clarity LIMS Web Interface, as shown in Figure 4. |
| • | Both of the attached files are placed on the Clarity LIMS server, in the /opt/gls/clarity/customextensions folder. |
| • | You will need to update the HOSTNAME global variable such that it points to your Clarity LIMS server. |
| • | The example code is provided for illustrative purposes only. It does not contain sufficient exception handling for use 'as is' in a production environment. |