Tumor Mutational Burden
DRAGEN tumor mutational burden (TMB) analysis step generates TMB metrics from the annotated small variant JSON file, the callability file generated from DRAGEN metrics and intermediate files generated from small variant calling. To remove germline variants from the TMB calculation, the software uses a combination of public database filtering and a post‑database filtering strategy that uses allele frequency information and variants in close proximity. Database filtering uses the GnomAD exome, genome, and 1000 genomes database.
The TMB is calculated using the
following equation:
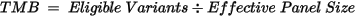
The eligible variants and effective panel size of the TMB calculation are detailed in the following table.
|
Eligible variants (numerator)
|
|
•
|
Variants in the coding region (RefSeq Cds). |
|
•
|
Variant frequency ≥ 0.2%. |
|
•
|
SNVs and Indels (MNVs excluded). |
|
•
|
Nonsynonymous and synonymous variants. |
|
•
|
Variants with cosmic count > 50 are excluded. |
|
•
|
Mutations in ASXL1, DNMT3A, PPM1D, and TET2 are excluded. |
|
•
|
Fragment-size based potential clonal hematopoiesis mutations are excluded. |
|
|
Effective panel size (denominator)
|
|
•
|
Total coding region with coverage ≥ 1000X. |
|
Outputs are captured in a *_tmb_trace.tsv file that contains the variants used in the TMB calculation, and a *.tmb.metrics.csv file that contains the TMB score calculation and configuration details.