DRAGEN Amplicon Pipeline
Amplicon sequencing is a highly targeted approach that enables you to analyze genetic variation in specific genomic regions. The ultradeep sequencing of PCR products (amplicons) allows you to efficiently identify and characterize variants. This method uses oligonucleotide probes designed to target and capture regions of interest, followed by next-generation sequencing (NGS).
The Amplicon Pipeline only supports DNA data. The Amplicon Pipeline uses the DRAGEN DNA Pipeline by including an additional step after mapping and aligning to soft-clip primers and rewrite alignments. If the target amplicon is found, DRAGEN tags each alignment with the target amplicon and performs soft-clipping the primer sequences. DRAGEN performs tagging by adding an XN:Z:<amplicon name> tag to the output BAM/CRAM record. Soft-clipping makes sure that the primer sequences do not contribute to the variant calls. The Amplicon Pipeline turns off duplicate marking because there are only a few unique start and end positions for fragments from an amplicon target due to the assay.
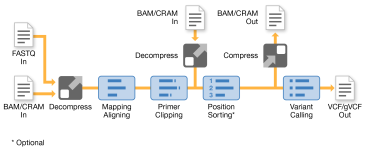
The DRAGEN Amplicon Pipeline requires an amplicon BED files and all input files required by the DRAGEN DNA pipeline. Each row in an amplicon BED file describes an amplicon target. The following fields are required.
|
Field |
Description |
|---|---|
|
chrom |
The name of the chromosome. |
|
chromStart |
The 0-based inclusive start position of the target, excluding the primer. |
|
chromEnd |
The 0-based exclusive end position of the target, excluding the primer. |
|
name |
The name of the amplicon target. |
|
gene |
[Optional] The gene ID. |
To use the amplicon pipeline, set --enable-dna-amplicon to true. Use --amplicon-target-bed to specify the path to your Amplicon BED file.
Only small variant calling is supported. To use small variant calling, set --enable-variant-calling to true and input the target small variant calling BED file using --vc-target-bed. Set the target bed file to the amplicon BED file. If calling somatic small variants, set --vc-use-somatic-hotspots to false.
By default the maximum amplicon primer length is set to 25. You can specify a different value using --amplicon-primer-length. The parameter affects whether an alignment is assigned to an amplicon target. If an alignment starts inside the primer region of the amplicon target, the alignment is assigned to the amplicon. For a properly paired alignment, both the alignment and the mate must come from the same amplicon target.
|-- primer --|-- amplicon target --|-- primer --|
---------- read ----------------->
<---------- read -----------------
The following is an example command line to run the DRAGEN Amplicon Pipeline with germline small variant calling.
dragen --enable-dna-amplicon true --enable-map-align=true --enable-sort=true --enable-map-align-output=true -r reference_genomes/Hsapiens/hg19_alt_aware/DRAGEN/8 --amplicon-target-bed=CancerHotSpot-v2.dna_manifest.20180509.bed --enable-variant-caller=true --vc-target-bed=CancerHotSpot-v2.dna_manifest.20180509.bed --fastq-file1=read1.fastq.gz --fastq-file2=read2.fastq.gz --RGSM NA12878 --RGID 1 --output-directory=/staging/out --output-file-prefix=NA12878