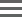Matched Normal Control Sample Post Somatic Filtering
|
Somatic Mode |
Filter ID |
Description |
|---|---|---|
|
Tumor-Normal |
noisy_normal |
More than three alleles are observed in the normal sample at allele frequency above 9.9%. |
|
Tumor-Normal |
alt_allele_in_normal |
Alt allele frequency in the normal sample is above 0.2 plus the maximum contamination tolerance. For solid tumor mode the value is 0. For liquid tumor mode the default value is 0.15. See vc-enable-vaf-ratio-filter for optional conditions. |
|
Tumor-Normal |
filtered_reads |
More than 90% of reads have been filtered out. |
|
Tumor-Normal |
no_reliable_supporting_read |
No reliable supporting read was found in the tumor sample. A reliable supporting read is a read supporting the alt allele with mapping quality ≥ 40, fragment length ≤ 10,000, basecall quality ≥ 25, and distance from start/end of read ≥ 5. |
|
Tumor-Normal |
strand_artifact |
Severe strand bias. Alt allele supported by at least four reads, but only on one strand. |
|
Tumor-Normal |
too_few_supporting_reads |
Variant is supported by < 3 reads in the tumor sample. |
|
Tumor-Normal |
non-homref_normal |
Normal sample genotype is not a homozygous reference. |
|
Tumor-Normal |
germline_risk |
Likelihood of allele being present in normal > 0.025. Enabled only when using --vc-enable-gatk-acceleration=true. |
|
Tumor-Normal |
artifact_in_normal |
TLOD of the normal read set (Normal artifact LOD) is > 0.0. Not called normal artifact if allele fraction in normal is much smaller than allele fraction in tumor (normalAlleleFraction < (0.1 * tumorAlleleFraction). Enabled only when using --vc-enable-gatk-acceleration=true. |
QUAL is not output in the somatic variant records. Instead, the confidence score is FORMAT/SQ.
##FORMAT=<ID=SQ,Number=1,Type=Float,Description="Somatic quality">
The field is specific to the sample. For the tumor samples, it quantifies the evidence that a somatic variant is present at a given locus.
If a normal sample is also available, the corresponding FORMAT/SQ value quantifies the evidence that the normal sample is a homozygous reference at a given locus.
GQ is not output in the somatic variant records, because DRAGEN does not test for multiple diploid genotype candidates. Instead, an ALT allele is considered as a candidate somatic variant. If tumor SQ > vc-sq-call-threshold (default is 3), then the FORMAT/GT for the tumor sample is hard-coded to 0/1, and the FORMAT/AF yields an estimate on the somatic variant allele frequency, which ranges anywhere within [0,1].
| • | If tumor SQ < vc-sq-call-threshold, the variant is not emitted in the VCF. |
| • | If tumor SQ > vc-sq-call-threshold but tumor SQ < vc-sq-filter-threshold, the variant is emitted in the VCF, but FILTER=weak_evidence. |
| • | If tumor SQ > vc-sq-call-threshold and tumor SQ > vc-sq-filter-threshold, the variant is emitted in the VCF and FILTER=PASS (unless the variant is filtered by a different filter). |
| • | The default vc-sq-filter-threshold is 17.5 for tumor-normal and 3.0 for tumor-only analysis. |
The following is an example somatic T/N VCF record. Tumor SQ > vc-sq-call-threshold but tumor SQ < vc-sq-filter-threshold, so the FILTER is marked as weak_evidence.
2 593701 . G A . weak_evidence
DP=97;MQ=48.74;SQ=3.86;NLOD=9.83;FractionInformativeReads=1.000
GT:SQ:AF:F1R2:F2R1:DP:SB:MB 0/0:9.83:33,0:0.000:14,0:19,0:33 0/1:3.86:61,3:0.047:29,2:32,1:64:35,26,0,3:39,22,1,2
The clustered-events penalty is an exception to the above rule for emitting variants. By default, the clustered-events penalty replaces the clustered-events filter in tumor-normal mode. Instead of applying a hard filter when too many events are clustered together, DRAGEN applies a penalty to the SQ scores of co-phased clustered events. Clustered events with weak evidence are no longer called, but clustered events with strong evidence can still be called. This is equivalent to lowering the prior probability of observing clustered co-phased variants. The penalty is applied after the decision to emit variants, so that penalized variants still appear in the VCF if their unpenalized score is high enough.