DRAGEN HLA Caller
DRAGEN includes a dedicated human leukocyte antigen (HLA) genotyper for calling HLA class I alleles with four-digit resolution. This resolution, also referred to as two-field resolution according to HLA nomenclature, determines the HLA protein. For more information on HLA nomenclature, see Nomenclature for factors of the HLA system¹.
You can enable HLA typing by setting --enable-hla flag to true. The following image represents an overview of the DRAGEN HLA Caller.
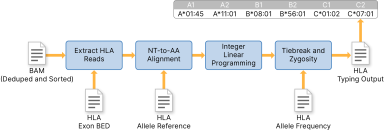
The DRAGEN HLA Caller performs four primary steps.
| 1. | Extracts reads from specified HLA loci, depending on the genome reference version used. See HLA Region BED Input File for more information on the required input file. |
| 2. | Uses amino-acids to perform alignment against specified HLA allele reference sequences. See HLA Allele Reference Input File for more information on the required reference input file. |
| 3. | Identifies a short list of possible candidate HLA alleles using integer linear programming (ILP). |
| 4. | Identifies the most plausible HLA genotypes by performing a tiebreak procedure and zygosity check. A specified population-level HLA allele frequency file is required. See HLA Allele Frequency Input File for more information. |
The following example command enables HLA typing, including the input defaults described below.
dragen \
--enable-hla=true \
--hla-bed-file=hla_exons_grch38.bed \
--hla-reference-file=hla_classI_ref_freq.fasta \
--hla-allele-frequency-file=hla_classI_allele_frequency.csv \
--hla-tiebreaker-threshold 0.97 \
--hla-zygosity-threshold 0.15 \
--output-directory={output_directory} \
--output-file-prefix={prefix} \
--enable-map-align=true \
--RGID=read_group_ID \
--RGSM=read_group_sample \
--ref-dir={reference_directory} \
--enable-map-align-output=true \
--enable-sort=true \
--enable-duplicate-marking=true \
-1 {fq1} \
-2 {fq2} \
You can use the HLA region BED input file to specify the region to extract HLA reads from. To specify an HLA region BED file, use --hla-bed-file. DRAGEN HLA Caller parses the input file for regions within the BED file, and then extracts reads accordingly to align with the HLA allele reference.
If using a human reference genome that DRAGEN can automatically detect, the HLA Caller chooses the corresponding BED file. If using a customized reference that cannot be automatically detected and a BED file is not specified, the HLA Caller provides a warning and uses all mapped reads instead.
The following is an example of a valid BED file.
chr6 29942554 29942627 hla_a 1 +
chr6 29942757 29943027 hla_a 2 +
chr6 29943268 29943544 hla_a 3 +
chr6 29944122 29944398 hla_a 4 +
chr6 29944500 29944617 hla_a 5 +
chr6 29945059 29945092 hla_a 6 +
chr6 29945234 29945282 hla_a 7 +
chr6 31357086 31357159 hla_b 1 -
chr6 31356688 31356958 hla_b 2 -
chr6 31356167 31356443 hla_b 3 -
chr6 31355317 31355593 hla_b 4 -
chr6 31355107 31355224 hla_b 5 -
chr6 31354633 31354666 hla_b 6 -
chr6 31354483 31354527 hla_b 7 -
chr6 31271999 31272072 hla_c 1 -
chr6 31271599 31271869 hla_c 2 -
chr6 31271073 31271349 hla_c 3 -
chr6 31270210 31270486 hla_c 4 -
chr6 31269966 31270086 hla_c 5 -
chr6 31269493 31269526 hla_c 6 -
chr6 31269338 31269386 hla_c 7 -
You can use the HLA allele reference file to specify the reference alleles to align against. To specify an HLA allele reference file, use the command line option --hla-reference-file. The input HLA reference file must be in FASTA format and contain the protein sequence separated into exons.
If --hla-reference-file is not specified, DRAGEN uses hla_classI_ref_freq.fasta from /opt/edico/config/. The reference HLA sequences are obtained from the IMGT/HLA database.
The following is an example of a valid reference file.
>A*01:01-E1
MAVMAPRTLLLLLSGALALTQTWAG
>A*01:01-E2
SHSMRYFFTSVSRPGRGEPRFIAVGYVDDTQFVRFDSDAASQKMEPRAPWIEQEGPEYWDQETRNMKAHSQTDRANLGTLRGYYNQSEDG
>A*01:01-E3
SHTIQIMYGCDVGPDGRFLRGYRQDAYDGKDYIALNEDLRSWTAADMAAQITKRKWEAVHAAEQRRVYLEGRCVDGLRRYLENGKETLQRTD
>A*01:01-E4
PPKTHMTHHPISDHEATLRCWALGFYPAEITLTWQRDGEDQTQDTELVETRPAGDGTFQKWAAVVVPSGEEQRYTCHVQHEGLPKPLTLRWE
>A*01:01-E5
LSSQPTIPIVGIIAGLVLLGAVITGAVVAAVMWRRKSSD
>A*01:01-E6
RKGGSYTQAAS
>A*01:01-E7
SDSAQGSDVSLTACKV
>A*01:03-E1
MAVMAPRTLLLLLSGALALTQTWAG
>A*01:03-E2
SHSMRYFFTSVSRPGRGEPRFIAVGYVDDTQFVRFDSDAASQKMEPRAPWIEQEGPEYWDQETRNMKAHSQTDRANLGTLRGYYNQSEDG
>A*01:03-E3
SHTIQMMYGCDVGPDGRFLRGYRQDAYDGKDYIALNEDLRSWTAADMAAQITKRKWEAVHAAEQRRVYLEGRCVDGLRRYLENGKETLQRTD
>A*01:03-E4
PPKTHMTHHPISDHEATLRCWALGFYPAEITLTWQRDGEDQTQDTELVETRPAGDGTFQKWAAVVVPSGEEQRYTCHVQHEGLPKPLTLRWE
>A*01:03-E5
LSSQPTIPIVGIIAGLVLLGAVITGAVVAAVMWRRKSSD
>A*01:03-E6
RKGGSYTQAAS
>A*01:03-E7
...
You can use the population-level HLA allele frequency file to break ties if one or more HLA allele produces the same or similar results. To specify an HLA allele frequency file, use the command line option --hla-allele-frequency-file. The input HLA allele frequency file must be in CSV format and contain the HLA alleles and the occurrence frequency in population.
If --hla-allele-frequency-file is not specified, DRAGEN automatically uses hla_classI_allele_frequency.csv from /opt/edico/config/. Population-level allele frequencies can be obtained from the Allele Frequency Net database.
The following is an example of a valid allele frequency file:
A*01:01,305
A*01:02,140
A*01:03,100
A*01:04N,13
A*01:06,58
A*01:07,17
A*01:08,14
A*01:09,25
...
You can use the following options to configure the HLA Caller.
| • | --hla-tiebreaker-threshold—If more than one allele has a similar number of reads aligned and there is not a clear indicator for the best allele, the alleles are considered as ties. The HLA Caller places the tied alleles into a candidate set for tie breaking based on the population allele frequency. If an allele has more than the specified fraction of reads aligned (normalized to the top hit), then the allele is included into the candidate set for tie breaking. The default value is 0.97. |
| • | --hla-zygosity-threshold—If the minor allele at a given locus has fewer reads mapped than a fraction of the read count of the major allele, then the HLA Caller infers homozygosity for the given HLA-I gene. You can use this option to specify the fraction value. The default value is 0.15. |
| • | --hla-min-reads—Set the minimum number of reads to align to HLA alleles to ensure sufficient coverage and perform HLA typing. The default value is 1000 and suggested for WES samples. If using samples with less coverage, you can use a lower threshold value. |
The DRAGEN HLA Caller generates HLA typing results with six class I alleles. The main output file can be called <prefix>.hla.tsv or <prefix>.hla.normal.tsv and <prefix>.hla.tumor.tsv. The file contains a header row with one column for each of the six alleles and a body row with the HLA types of each allele at four-digit resolution.
The following is an example output file.
A1 A2 B1 B2 C1 C2
A*26:01 A*29:02 B*44:02 B*44:03 C*05:01 C*16:01
The HLA Caller generates the following additional HLA files that you can use to assess the intermediate steps of HLA typing.
| • | <prefix>.freq_tiebreaking_candidates.tsv—Contains the candidate allele from the tie breaking process. The file includes the number of exclusive reads and population frequency of the candidate alleles. |
| • | <prefix>.hla_metrics.csv—Contains the number of reads extracted from the HLA region, allele set selected by ILP, the number of reads explained by ILP selected allele set, and the number of exclusive reads with which zygosity is determined. |
| • | Using alt-aware references are not recommended because the read extraction step causes reduced accuracy. You can achieve more accurate HLA typing using masked alt-aware references, where regions in the alternative haplotypes with a high similarity to the primary assemble are masked with Ns. |
| • | DRAGEN only supports HLA typing for class I HLA genes. |
The HLA Caller accepts standard input files in FASTQ or BAM format, as well as an HLA region BED file, HLA allele reference file, and HLA frequency file. If not specified, the HLA Caller uses the files located in src/config/hla.
The following example command line uses FASTQ file inputs and the default options.
dragen \
--enable-hla=true \
--enable-map-align=true \
--enable-sort=true \
--enable-duplicate-marking=true \
--output-directory={output_directory} \
--output-file-prefix={prefix} \
--ref-dir={reference_directory} \
--RGID={read_group_ID} \
--RGSM={read_group_sample} \
-1 {fq1} \
-2 {fq2} \
The following example command line uses BAM file inputs and the default options.
dragen \
--enable-hla=true \
--enable-map-align=true \
--enable-sort=true \
--enable-duplicate-marking=true \
--output-directory={output_directory} \
--output-file-prefix={prefix} \
--bam-input {bam} \
--ref-dir={reference_directory} \
The following example command line uses tumor-normal paired file inputs and the default options.
dragen \
--enable-hla=true \
--enable-map-align=true \
--enable-sort=true \
--output-directory={output_directory} \
--output-file-prefix={prefix} \
--ref-dir={reference_directory} \
--tumor-fastq1 {tumor_fq1} \
--tumor-fastq2 {tumor_fq2} \
--RGID-tumor={tumor_group_ID} \
--RGSM-tumor={tumor_group_sample} \
-1 {normal_fq1} \
-2 {normal_fq2} \
--RGID={normal_group_ID} \
--RGSM={normal_group_sample} \
¹Marsh SG, et al. Nomenclature for factors of the HLA system, 2010. Tissue Antigens. 2010 75:291-455.