DRAGEN Methylation Pipeline
The epigenetic methylation of cytosine bases in DNA can have a dramatic effect on gene expression, and bisulfite sequencing is the most common method for detecting epigenetic methylation patterns at single-base resolution. This technique involves chemically treating DNA with sodium bisulfite, which converts unmethylated cytosine bases to uracil, but does not alter methylated cytosines. Subsequent PCR amplification converts any uracils to thymines.
A bisulfite sequencing library can either be nondirectional or directional. For nondirectional, each double-stranded DNA fragment yields four distinct strands for sequencing, post-amplification, as shown in the following figure:
Nondirectional Bisulfite Sequencing
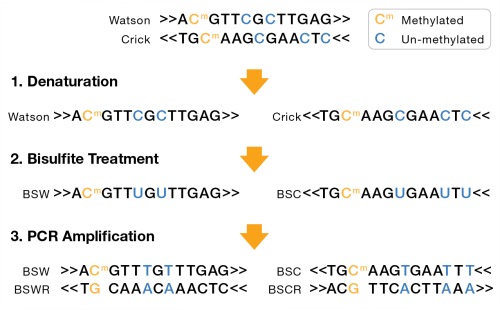
| – | Bisulfite Watson (BSW), reverse complement of BSW (BSWR), |
| – | Bisulfite Crick (BSC), reverse complement of BSC (BSCR) |
For directional libraries, the four strand types are generated, but adapters are attached to the DNA fragments such that only the BSW and BSC strands are sequenced (Lister protocol). Less commonly, the BSWR and BSCR strands are selected for sequencing (eg, PBAT).
BSW and BSC strands:
| • | A, G, T: unchanged |
| • | Methylated C remains C |
| • | Unmethylated C converted to T |
BSWR and BSCR strands:
| • | Bases complementary to original Watson/Crick A, G, T bases remain unchanged. |
| • | G complementary to original Watson/Crick methylated C remains G. |
| • | G complementary to original Watson/Crick unmethylated C becomes A. |
To run DRAGEN methylation pipeline, you need to build a methylation-specific hash table. Both multipass and single-pass options require methylation hash tables. The type of hash table must match the method used during alignment (methylation-mapping-implementation: single-pass| multi-pass). For more information on multipass and single-pass methods, see Mapping Method Options.
If using the single-pass method, make sure that your hash table meets the following requirements:
| • | Set methylation-mapping-implementation: single-pass. |
| • | Set ht-methylated-combined=true to generate a combined hash table in the methyl_converted subdirectory. |
Each contig in the original FASTA appears twice in the combined reference genome files, one time with each conversion type.
If using the multipass method, make sure that your hash table meets the following requirements:
| • | Set methylation-mapping-implementation: multi-pass. |
| • | Set ht-methylated=true to generate the CT_converted and GA_converted subdirectories. |
The subdirectories contain the genome index for C to T and G to A conversion.
You can set ht-methylated-combined=true and ht-methylated=true together to create a hash table directory that can be used for both methylation-mapping methods.
The following is an example command line for a single-pass hash table.
dragen --build-hash-table true \
--output-directory=sample.output.directory \
--ht-reference=sample.input.fa \
--ht-num-threads 40 \
--ht-methylated=true \
--ht-methylated-combined=true \
--ht-seed-len 27
The --ht-seed-len 27 option is important for optimal results. The --ht-num-threads option enables multiple numbers of threads for faster building.