
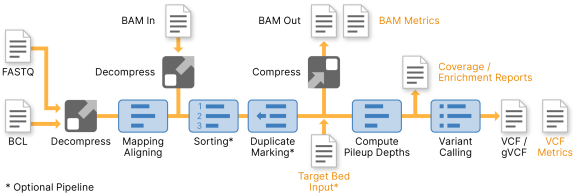
QC Metrics and Coverage/Callability Reports
DRAGEN generates pipeline-specific metrics coverage reports during each run. There are four different groups of metrics that are generated at different stages of the pipeline:
| • | Mapping and Aligning metrics |
| • | VCF metrics |
| • | Duration (or run time) metrics |
| • | Coverage (or enrichment) metrics and reports |
The mapping/aligning metrics, VCF metrics, Duration metrics, and a subset of available coverage reports are autogenerated and do not require any activation or specific commands. Additional coverage metrics can be enabled, and additional coverage regions can be specified.
DRAGEN performs metric calculation during analysis so that it does not impact the run time.
Generation of Metrics and Reports
The QC metrics are printed to the standard output and CSV files are written to the run output directory.
| • | <output prefix>.mapping_metrics.csv |
| • | <output prefix>.vc_metrics.csv |
| • | <output prefix>.<coverage region prefix>_coverage_metrics.csv |
| • | <output prefix>.time_metrics.csv |
| • | <output prefix>.<other coverage reports>.csv |
|
Section |
RG/Sample |
Metric |
Count/Ration/Time |
Percentage/Seconds |
|---|---|---|---|---|
|
MAPPING/ALIGNING SUMMARY |
|
Total input reads |
816360354 |
|
|
MAPPING/ALIGNING SUMMARY |
|
Number of duplicate reads (marked not removed) |
15779031 |
1.93 |
|
... |
|
|
|
|
|
MAPPING/ALIGNING PER RG |
RGID_1 |
Total reads in RG |
816360354 |
100 |
|
MAPPING/ALIGNING PER RG |
RGID_1 |
Number of duplicate reads (marked) |
15779031 |
1.93 |
|
... |
|
|
|
|
|
VARIANT CALLER SUMMARY |
|
Number of samples |
1 |
|
|
VARIANT CALLER SUMMARY |
|
Reads Processed |
738031938 |
|
|
... |
|
|
|
|
|
VARIANT CALLER PREFILTER |
SAMPLE_1 |
Total |
4918287 |
100 |
|
VARIANT CALLER PREFILTER |
SAMPLE_1 |
Biallelic |
4856654 |
98.75 |
|
... |
|
|
|
|
|
COVERAGE SUMMARY |
|
Aligned bases in genome |
14847587 |
100.00 |
|
COVERAGE SUMMARY |
|
Average alignment coverage over genome |
0.03 |
|
|
... |
|
|
|
|
|
RUN TIME |
|
Time loading reference |
00:18.6 |
18.65 |
|
RUN TIME |
|
Time aligning reads |
19:24.4 |
1164.42 |
Mapping and aligning metrics, such as the metrics computed by the Samtools Flagstat command, are available on an aggregate level (over all input data), and on a per read group level. Unless explicitly stated, the metrics units are in reads (ie, not in terms of pairs or alignments).
|
Metric |
Description |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Total input reads |
Total number of reads in the input FASTQ files. |
|||||||||||||||
|
Number of duplicate marked reads |
Reads marked as duplicates as a result of the --enable-duplicate-marking option being set to true. |
|||||||||||||||
|
Number of duplicate marked and mate reads removed |
Reads marked as duplicates, along with any mate reads that are removed when the --remove-duplicates option is set to true. |
|||||||||||||||
|
Number of unique reads |
Total number of reads minus the duplicate marked reads. |
|||||||||||||||
|
Reads with mate sequenced |
Number of reads with a mate. |
|||||||||||||||
|
Reads without mate sequenced |
Total number of reads minus number of reads with mate sequenced. |
|||||||||||||||
|
QC-failed reads |
Reads that did not pass platform/ vendor quality checks (SAM flag 0x200). |
|||||||||||||||
|
Mapped reads |
Total number of mapped reads minus number of unmapped reads. |
|||||||||||||||
|
Number of unique and mapped reads |
Number of mapped reads minus number of duplicate marked reads. |
|||||||||||||||
|
Unmapped reads |
Total number of reads that could not be mapped. |
|||||||||||||||
|
Singleton reads |
Number of reads where the read could be mapped, but the paired mate could not be read. |
|||||||||||||||
|
Paired reads |
Count of reads in which both reads in the pair are mapped. |
|||||||||||||||
|
Properly paired reads |
Both reads in the pair are mapped and fall within an acceptable range from each other based on the estimated insert length distribution. |
|||||||||||||||
|
Not properly paired reads (discordant) |
The number of paired reads minus the number of properly paired reads. |
|||||||||||||||
|
Paired reads mapped to different chromosomes |
The number of reads with a mate, where the mate was mapped to a different chromosome. |
|||||||||||||||
|
Paired reads mapped to different chromosomes (MAPQ >= 10) |
The number of reads with a MAPQ > 10 and with a mate, where the mate was mapped to a different chromosome. |
|||||||||||||||
|
Reads with indel R1 |
The percentage of R1 reads containing at least 1 indel. |
|||||||||||||||
|
Reads with indel R2 |
The percentage of R2 reads containing at least 1 indel. |
|||||||||||||||
|
Soft-clipped bases R1 |
The percentage of bases in R1 reads that are soft-clipped. |
|||||||||||||||
|
Soft-clipped bases R2 |
The percentage of bases in R2 reads that are soft-clipped. |
|||||||||||||||
|
Mismatched bases R1 |
The number of mismatched bases on R1, which is the sum of SNP count and indel lengths. The metric does not count anything within soft clipping or RNA introns. The metric also does not count a mismatch if either the reference base or read base is N. |
|||||||||||||||
|
Mismatched bases R2 |
The number of mismatched bases on R2, which is the sum of SNP count and indel lengths. The metric does not count anything within soft clipping or RNA introns. The metric also does not count a mismatch if either the reference base or read base is N. |
|||||||||||||||
|
Mismatched bases R1 (excluding indels) |
The number of mismatched bases on R1. The indels lengths are ignored. It does not count anything within soft clipping or RNA introns. The metric also does not count a mismatch if either the reference base or read base is N. |
|||||||||||||||
|
Mismatched bases R2 (excluding indels) |
The number of mismatched bases on R2. The indels lengths are ignored. The metric does not count anything within soft clipping or RNA introns. The metric also does not count a mismatch if either the reference base or read base is N. |
|||||||||||||||
|
Q30 Bases |
The total number of bases with a BQ ≥ 30. |
|||||||||||||||
|
Q30 Bases R1 |
The total number of bases on R1 with a BQ ≥ 30. |
|||||||||||||||
|
Q30 Bases R2 |
The total number of bases on R2 with a BQ >= 30. |
|||||||||||||||
|
Q30 Bases (excluding dups and clipped bases) |
The number of bases on nonduplicate and nonclipped bases with a BQ ≥ 30. |
|||||||||||||||
|
Histogram of reads map qualities |
|
|||||||||||||||
|
Total alignments |
Total number of loci reads aligned to with > 0 quality. |
|||||||||||||||
|
Secondary alignments |
Number of secondary alignment loci. |
|||||||||||||||
|
Supplementary (chimeric) alignments |
A chimeric read is split over multiple loci (possibly due to structural variants). One alignment is referred to as the representative alignment. The other are supplementary. |
|||||||||||||||
|
Estimated read length |
Total number of input bases divided by the number of reads. |
|||||||||||||||
|
Histogram |
||||||||||||||||
|
PCT of bases aligned that fell inside the interval region |
Number of bases inside the interval region and the target region divided by the total number of bases aligned. |
|||||||||||||||
|
Estimated sample contamination |
The estimated fraction of reads in a sample that may be from another human source. |
The DRAGEN cross-sample contamination module uses a probabilistic mixture model to estimate the fraction of reads in a sample that may be from another human source. DRAGEN supports separate modes for germline and somatic samples.
The germline model, like VerifyBamID, assumes that a sample can be modeled as a DNA mixture from 2 or more individuals. Pileup analysis investigates loci where variants are common in the general population. Variants with high allele frequencies are likely to be real germline variants in the individual of interest, while low allele frequency variants are likely noise or germline variants from a contaminating sample B. The probabilistic mixture model accounts for noise and tries to detect consistent allele frequency distributions.
For example, if the pileups show consistent low allele frequencies of 1% or 2%, then the mixture model will likely infer 2% contamination from sample B, where the 1% and 2% AF variants correspond to heterozygous and homozygous germline calls in sample B.
The germline cross-contamination metric is enabled by using the following setting and pointing a VCF that includes marker sites (RSIDs) with population allele frequencies that are close to 0.5.
--qc-cross-cont-vcf /opt/edico/config/sample_cross_contamination_resource_[hg19 or GRCh37 or GRCh38].vcf
The metric value is printed as a fraction, so a value of 0.011 represents 1.1% contamination from another sample.
MAPPING/ALIGNING SUMMARY Estimated sample contamination 0.011
Variant calling precision, especially somatic variant calling precision, can be significantly affected by cross-sample contamination. To safely use a sample, the cross-sample contamination level should be significantly lower than the minimum allele frequencies of interest. For example, if a sample has 1% contamination, then it may be pertinent to ignore all variants with less than 5% AF. The sample cross-contamination metric saturates close to 30% contamination.
In somatic tumor-normal mode, the mapping and aligning metrics are generated separately for the tumor and normal samples, with each line beginning with TUMOR or NORMAL to indicate the sample. The metrics for the tumor sample are output first, followed by the metrics for the normal sample. Metrics per read group are also separated into tumor and normal read groups.
In somatic tumor-only mode, the mapping and aligning metrics follow the same conventions as germline mode, without metrics labeled as TUMOR or NORMAL.
The generated variant calling metrics are similar to the metrics computed by RTG vcfstats. Metrics are reported for each sample in multi sample VCF and gVCF files. Based on the run case, metrics are reported either as standard VARIANT CALLER or JOINT CALLER. Metrics are reported both for the raw (PREFILTER) and hard filtered (POSTFILTER) VCF file.
Panel of Normals (PON) and COSMIC filtered variants are counted as PASS variants in the POSTFILTER VCF metrics. These PASS variants can cause higher than expected variant counts in the POSTFILTER VCF metrics.
|
Metric |
Description |
|---|---|
|
Number of samples |
Number of samples in the population/ joint VCF. |
|
Reads Processed |
The number of reads used for variant calling, excluding any duplicate marked reads and reads falling outside of the target region. |
|
Total |
The total number of variants (SNPs + MNPs + indels). |
|
Biallelic |
Number of sites in a genome that contains two observed alleles. The reference is counted as one allele, which allows for one variant allele. |
|
Multiallelic |
Number of sites in the VCF that contain three or more observed alleles. The reference is counted as one, which allows for two or more variant alleles. |
|
SNPs |
A variant is counted as an SNP when the reference, allele 1, and allele 2 are all length 1. |
|
Insertions (Hom) |
Number of variants that contains homozygous insertions. |
|
Insertions (Het) |
Number of variants where both alleles are insertions, but not homozygous. |
|
Deletions (Het) |
Number of variants that contains homozygous deletions. |
|
INDELS (Het) |
Number of variants where genotypes are either [insertion+deletion], [insertion+SNP], or [deletion+SNP]. |
|
De Novo SNPs |
De novo marked SNPs with DQ > 0.05. Set the --qc-snp-denovo-quality-threshold option to the required threshold. The default is 0.05. |
|
De Novo INDELs |
De novo marked indels with DQ values > 0.02. This DQ threshold can be specified by setting the --qc-indel-denovo-quality-threshold option to the required DQ threshold. The default is 0.02. |
|
De Novo MNPs |
De novo marked SNPs with DQ > 0.05. Set the --qc-snp-denovo-quality-threshold to the required threshold. The default is 0.05. |
|
(Chr X SNPs)/(Chr Y SNPs) ratio in the genome (or the target region) |
Number of SNPs in chromosome X (or in the intersection of chromosome X with the target region) divided by the number of SNPs in chromosome Y (or in the intersection of chromosome Y with the target region). If there was no alignment to either chromosome X or chromosome Y, this metric shows as NA. |
|
SNP Transitions |
An interchange of two purines (A<->G) or two pyrimidines (C<->T). |
|
SNP Transversions |
An interchange of purine and pyrimidine bases Ti/Tv ratio: ratio of transitions to transitions. |
|
Heterozygous |
Number of heterozygous variants. |
|
Homozygous |
Number of homozygous variants. |
|
Het/Hom ratio |
Heterozygous/ homozygous ratio. |
|
In dbSNP |
Number of variants detected that are present in the dbSNP reference file. If no dbSNP file is provided via the --bsnp option, then both the In dbSNP and Novel metrics show as NA. |
|
Novel |
Total number of variants minus number of variants in dbSNP. |
|
Percent Callability |
Available in germline and somatic modes with gVCF output. The percentage of non-N reference positions having a PASSing genotype call. Multiallelic variants are not counted. Deletions are counted for all the deleted reference positions only for homozygous calls. Only autosomes and chromosomes X, Y, and M are considered. To produce this metric for non-human references, set --qc-callability-autosome-contigs to specify the autosome contig names. Optionally, --qc-callability-xym-contigs allows setting X, Y and M contig names. |
|
Percent Autosome Callability |
Only autosomes are considered. To produce this metric for non-human references, set --qc-callability-autosome-contigs to specify the autosome contig names. |
|
Percent QC Region Callability in Region i (i is equivalent to regions 1, 2, or 3) |
Available if callability for custom regions is requested via the --qc-coverage-region-i option and the callability output is specified with --qc-coverage-reports-i. All contigs are considered. Setting --qc-callability-autosome-contigs enables outputting this metric for non-human references. |
When the germline small variant caller is executed, DRAGEN calculates a per het/hom ratio per contig. DRAGEN reports the ratios for both the raw (PREFILTER) and hard-filtered (POSTFILTER) VCF. The metrics are output to the .vc_hethom_ratio_metrics.csv file. The file contains the following values for each primary contig processed.
| • | Contig |
| • | Number of heterozygous variants |
| • | Number of homozygous variants |
| • | Het/Hom ratio |
The following example shows a section of the metrics.
VARIANT CALLER POSTFILTER,HG04070,1 Heterozygous,185733
VARIANT CALLER POSTFILTER,HG04070,1 Homozygous,182928
VARIANT CALLER POSTFILTER,HG04070,1 Het/Hom ratio,1.015
VARIANT CALLER POSTFILTER,HG04070,2 Heterozygous,203946
VARIANT CALLER POSTFILTER,HG04070,2 Homozygous,174294
VARIANT CALLER POSTFILTER,HG04070,2 Het/Hom ratio,1.170
VARIANT CALLER POSTFILTER,HG04070,3 Heterozygous,192861
VARIANT CALLER POSTFILTER,HG04070,3 Homozygous,130087
VARIANT CALLER POSTFILTER,HG04070,3 Het/Hom ratio,1.483
VARIANT CALLER POSTFILTER,HG04070,4 Heterozygous,178389
VARIANT CALLER POSTFILTER,HG04070,4 Homozygous,157062
VARIANT CALLER POSTFILTER,HG04070,4 Het/Hom ratio,1.136
You can use the het/hom ratio values as an indication of whole chromosome uniparental disomy (UPD). UPD of certain chromosomes are associated with genetic syndromes known as imprinting disorders. Whole chromosome UPD have het/hom ratios close to 0.0. Ranges vary, but are usually between 1.0–2.0. Make sure you are interpreting het/hom ratios in the context of your own sequencing data.
The duration metrics section includes a breakdown of the run duration for each process. For example, the following metrics are generated for the mapper and variant caller pipeline:
| • | Time loading reference |
| • | Time aligning reads |
| • | Time sorting and marking duplicates |
| • | Time DRAGStr calibration |
| • | Time partial reconfiguration |
| • | Time variant calling |
| • | Total run time |
