Multiomics Pipeline
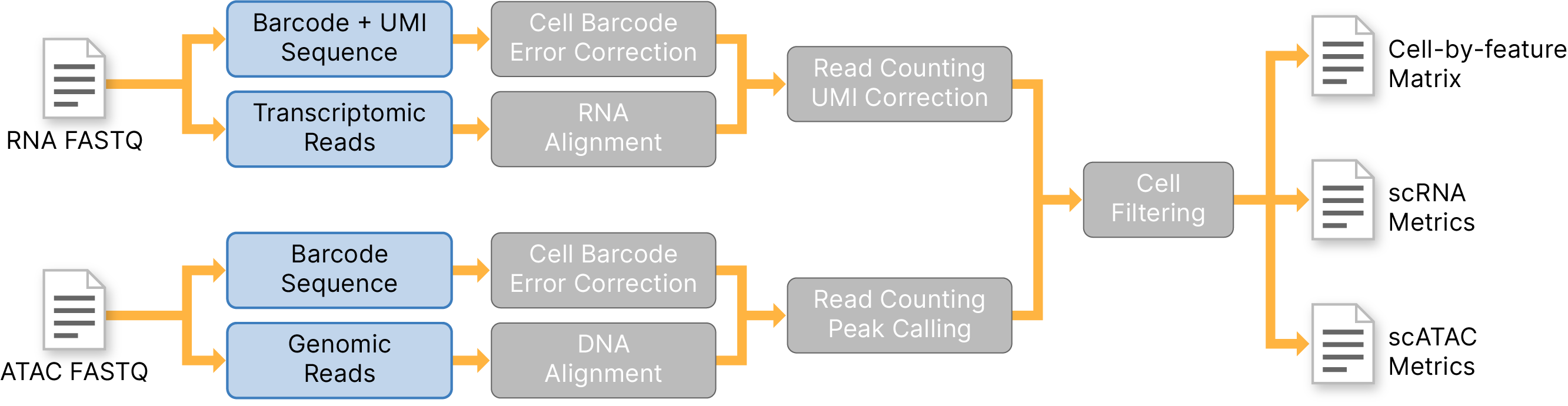
The DRAGEN Single-Cell Multiomics (scRNA + scATAC) Pipeline can process data sets from single-cell RNA-Seq and ATAC-seq reads to a cell-by-feature count matrix.
The pipeline is compatible with library designs that have:
|
•
|
Single-cell RNA: One read in a fragment match to a transcript and the other containing a cell-barcode and UMI |
|
•
|
Single-cell ATAC: Two reads matching to the genome and the third one containing cell-barcode |
The pipeline includes the following functions:
|
–
|
RNA-Seq (splice-aware) alignment and matching to annotated genes for the transcript reads |
|
•
|
Cell-barcode (both RNA- and ATAC-seq) and UMI (RNA-Seq only) error correction for the barcode read |
|
–
|
UMI counting per cell and gene to measure gene expression |
|
–
|
Fragment counting per cell and peak to measure chromatin accessibility |
|
•
|
Sparse matrix output and QC metrics |