
Iterative gVCF Genotyper Analysis
Iterative gVCF Genotyper Analysis offers an iterative workflow to aggregate new samples into an existing cohort. The iterative workflow allows users to incrementally aggregate new batches of samples with existing batches, without having to redo the analysis from scratch across all samples every time when new samples are available. The workflow takes single sample gVCF files as input, and can be performed in a step-by-step mode if multiple batches of samples are available, or end-to-end mode, if only a single batch of samples is available. Multi-sample gVCF files output from the Pedigree Caller are also accepted as input.
Workflow steps
| 1. | gVCF aggregation |
Users can use iterative gVCF Genotyper to aggregate a batch of gVCF files into a cohort file and a census file. The cohort file is a condensed data format to store gVCF data in multiple samples, similar to a multisample gVCF. The census file stores summary statistics of all the variants and hom-ref blocks among samples in the cohort. When a large number of samples are available, users can divide samples into multiple batches each with a similar sample size (eg 1000 samples), and repeat Step 1 for every batch. If force genotyping was enabled for any input file, any ForceGT calls that are not also called by the variant caller will be ignored.
| 2. | Census aggregation |
After all per batch census files are generated, users can aggregate them into a single global census file. This step scales to aggregate thousands of batches, in a much more efficient way compared to aggregating gVCFs from all batches. When a new batch of samples becomes available users only need to perform Step 1 on the batch, aggregate the census file from the batch with the global census file from all previous batches, and generate an updated global census file.
| 3. | msVCF generation |
When a global census file is updated with new variant sites discovered and/or variant statistics updated at existing variant sites, the user can take per-batch cohort file, per-batch census file, and the global census file as input, and generate a multisample VCF for one batch of samples. The output multisample VCF contains the variants and alleles discovered in all samples from all batches, and also includes global statistics such as allele frequencies, the number of samples with or without genotypes, and the number of samples without coverage. Similar statistics among samples in the batch are also included. This step can be repeated for every batch of samples, and the number of records in each output multisample VCF is the same across all batches.
To facilitate parallel processing on distributed compute nodes the user can choose to split the genome into shards of equal size for each step, and process each shard using one instance of iterative gVCF Genotyper on each compute node. See option --shard.
There is a special treatment of alternative or unaligned contigs when the --shard option is enabled: all contigs that are not autosomes, X, Y or chrM are included in the last shard. No other contigs will be assigned to the last shard. The mitochondrial contig will always be on its own in the second to last shard.
If a combined msVCF of all batches is required, an additional step should run separately to merge all of the batch msVCF files into a single msVCF containing all samples.
|
--enable-gvcf-genotyper-iterative |
Set to true to run the iterative gVCF genotyper (always required). |
||||||
|
--ht-reference |
The file containing the reference sequence in FASTA format (always required). |
||||||
|
--output-directory |
The output directory (always required). |
||||||
|
--output-file-prefix |
The prefix used to label all output files (optional, default value dragen). |
||||||
|
--shard |
Use this option to process only a portion (shard) of the genome, when distributing the work across multiple compute nodes. Provide the index (1-based) on the shard to process and the total number of shards, in the format n/N (eg 1/50 = shard 1 of 50 shards). To facilitate concurrent processing within each job, the shard will by default be split into 10x the number of available threads. |
||||||
|
--gg-regions |
if the --shard option is not given, use this option to run iterative gVCF genotyper only for a subset of regions in the genome. The same regions must be used for each step.
|
||||||
|
--gg-regions-bed |
If a path to a BED file is provided, it will limit the iterative gVCF Genotyper processing to the genome specified regions. This option differs from --gg-regions-bed in that, if the number of regions exceeds 10 times the number of available threads, for example exome data, they will not necessarily be processed by independent threads, making the option faster and compatible with sharding. In this situation there will only take effect on step 1 or end-to-end mode. |
||||||
|
--gg-discard-ac-zero |
If set to true, the gVCF Genotyper does not print variant alleles that are not called (hom-ref genotype) in any sample. The default value is true. |
||||||
|
--gg-remove-nonref |
Removes the <NON_REF> symbolic allele from the output of gVCF Genotyper. This option should be used to support downstream tools that cannot process VCF lines with <NON_REF> or to generate more concise msVCFs. This option needs to be enabled in step 1. The default is false. |
||||||
|
--gg-vc-filter |
Discard input variants that failed filters in the upstream caller. The default is false. Affected records will have their genotype set to hom-ref and the filter string "ggf" added to FORMAT/FT. |
||||||
|
--gg-hard-filter |
Specifies a filtering expression to be applied to the output msVCF records. See "msVCF hard filtering" below. The default is to apply no filters. |
||||||
|
--gg-skip-filtered-sites |
Omits msVCF records that fail the given hard filter. The default is false. |
|
--gvcfs-to-cohort-census |
Set to true to aggregate gVCF files from one batch of samples into a cohort file and a census file. |
|
--variant-list |
The path to a file containing a list of input gVCF files, with the path to each file on a separate line. |
|
--variant |
if --variant-list is not given, use this option for each input gVCF file. |
|
--aggregate-censuses |
Set to true to aggregate a list of per batch census files into a global census file. |
|
--input-census-list |
The path to a file containing a list of input per batch census files (from Step1), with the path to each file on a separate line. |
|
--generate-msvcf |
Set to true to generate a multisample VCF for one batch of samples. |
|
--input-cohort-file |
The path to the per batch cohort file (from Step 1). |
|
--input-census-file |
The path to the per batch census file (from Step 1). |
|
--input-global-census-file |
The path to the global census file (from Step 2). |
|
--variant-list |
The path to a file containing a list of input gVCF files, with the path to each file on a separate line. |
|
--variant |
If --variant-list is not given, use this option for each input gVCF file. |
|
--merge-batches |
Set to true to merge msVCF files for a set of batches. |
|
--input-batch-list |
The path to a file containing a list of msVCF files to be merged, with the path to each file on a separate line. All the files listed must have been generated from the same global census file and all batches pertaining to that global census must be included in the merge. |
|
--gg-enable-indexing |
Set to true to generate a tabix index for the merged msVCF (default false). |
Mimalloc is a custom memory allocation library that can yield significant speed-ups in the iterative gVCF Genotyper workflow. In some deployments, eg cloud, it is automatically and seamlessly used but in other contexts it requires special user intervention to be activated, as at present it cannot be included in standard DRAGEN by default.
The convenience script mi_dragen.sh is provided, that loads the bundled library and can be transparently used in the same way as the DRAGEN executable. It is only intended and supported for use with the iterative gVCF Genotyper component, although it can in principle be applied for any other DRAGEN workflow.
The use of Mimalloc for other purposes is known to potentially lead to undesirable memory overuse and is not recommended. Using Mimalloc for other purposes is at your own risk.
The output of gVCF Genotyper is a multi-sample VCF (msVCF) that contains metrics computed for all samples in the cohort.
The msVCF can become a very large file with increasing cohort size. In some cases, the file might need more storage than can be allocated by VCF parsers. This is caused by VCF entries such as FORMAT/PL which store a value for each combination of alleles. FORMAT/PL was replaced with a tag FORMAT/LPL which stores a value only for the alleles that occur in the sample. The FORMAT/LAA field lists 1-based indices of the alternate alleles that occur in the current sample.
When processing mitochondrial variant calls, which may contain separate records for each allele, iterative gVCF Genotyper processing differs in the following ways:
| • | Only the record with the highest FORMAT/AF sum is kept. |
| • | The FORMAT/AF field will be additionally collected, and used to generate the FORMAT/LAF field in the output msVCF |
The value displayed in the QUAL column of the msVCF is the maximum of the input QUAL values for the site across the global cohort. The QUAL value will be missing if any of the batch census files used to create the global census were generated with a version of DRAGEN earlier than v4.2.
The Hardy-Weinberg Equilibrium (HWE) states that, given certain conditions, genotype and allele frequencies should remain constant between generations. Deviations from HWE can results from violations of the underlying HWE assumptions in the population, non-random sampling or may be artifacts of variant calling. Adherence to HWE can be assessed by comparing the observed frequencies of genotypes to those expected under HWE given the observed allele counts.
Iterative gVCF Genotyper (iGG) offers several metrics for assessing adherence to HWE. It calculates both allele-wise and site-wise HWE P-values, an allele-wise excess heterozygosity (ExcHet) P-value and the site-wise inbreeding coefficient (IC). These metrics are calculated only for diploid sites and missing values are excluded from the calculations. These values are included as fields in the INFO column of the output msVCF file. Both batch-wise and global values are included, where the field names for the global values are prefixed with G.
|
Metric |
Description |
Scope |
Number of values |
|
HWE |
Hardy-Weinberg Equilibrium P-value |
Allele-wise |
One for each alt allele |
|
ExcHet |
Excess Heterozygosity P-value |
Allele-wise |
One for each alt allele |
|
HWEc2 |
Hardy-Weinberg Equilibrium P-value |
Site-wise |
1 |
|
IC |
Inbreeding Coefficient |
Site-wise |
1 |
Care should be taken when interpreting these metrics for small cohorts and/or low frequency alleles, as small changes in inputs can lead to large changes in their values. Further, violations of the underlying HWE assumptions (such as inbreeding), and non-random sampling (such as the presence of consanguineous samples), can adversely affect results, making identification of poorly called variants more difficult.
Where it is not possible to calculate the metric, they are represented as missing (ie, ".") in the msVCF file. This can vary between the metrics, but may occur if non-diploid genotypes are encountered, if there is only one allele present at a site, or if no samples are genotyped at a site.
iGG calculates allele-wise HWE and the ExcHet P-values. The values are calculated using the exact-conditional method described in Am J Hum Genet. 2005 May; 76(5): 887–893. The implementation does not use a mid P-value correction.
For HWE a P-value of ≈ 1 suggests that the distribution of heterozygotes and homozygotes is close to that expected under HWE, while a P-value of ≈ 0 suggests a deviation from it. For ExcHet a P-value of ≈ 0.5 suggests that the number of heterozygotes is close to the number expected under HWE, while a value ≈ 1 suggests that there are more heterozygotes than expected and a value ≈ 0 suggests that there are fewer heterozygotes than expected.
For a bi-allelic site the HWE P-values is based on the numbers of homozygotes and heterozygotes comparing the observed to expected. For a multi-allelic site, P-values are calculated per ALT allele as if it were bi-allelic. Genotypes composed of only the ALT allele being considered are counted as alternative homozygous, any other genotype containing a copy of the ALT allele being considered are counted as a heterozygous, and any genotype with no copies of the ALT allele being considered are counted as reference homozygous (this may include genotypes containing other ALT alleles).
Iterative gVCF Genotyper calculates a site-wise HWE P-value. The value is calculated using the Pearson's chi-squared method, comparing the genotype counts expected under HWE to those observed. The chi-squared test statistic is calculated as:
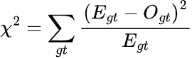
where the summation gt is over is over all genotypes possible at the site given the alleles present, 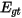 and
and 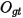 are the expected and observed counts for genotype gt respectively. From the chi-squared test statistic the P-value is then calculated from a chi-squared distribution where the number of degrees of freedom is the number of possible genotypes minus the number of alleles, which is
are the expected and observed counts for genotype gt respectively. From the chi-squared test statistic the P-value is then calculated from a chi-squared distribution where the number of degrees of freedom is the number of possible genotypes minus the number of alleles, which is
dof = n(n-1)/2
where n is the number of alleles.
The batch-wise value uses only the alleles present in the batch. Alleles with AC=0 are not included in the calculation.
A P-value of ≈ 1 suggests that the distribution of heterozygotes and homozygotes is close to that expected under HWE, while a P-value of ≈ 0 suggests a deviation from it.
iGG calculates the inbreeding coefficient (IC) (sometimes called the Fixation index and denoted by F). It is defined as the proportion of the population that is inbred. The value of IC can be estimated by looking at the observed number of heterozygotes in comparison to the number expected under HWE:
IC = 1 - O(het) / E(het),
where O(het) and E(het) are the observed and expected number of heterozygotes in the cohort, respectively. Although initially conceived for studying inbreeding and defined as a non-negative value, it is also commonly used to look for deviations from HWE and can take values in the range [-1, 1].
Values of IC ≈ 0 suggest that the cohort is in HWE. Negative values suggest an excess of heterozygosity and a deviation from HWE, which can be symptomatic of poor variant calling. Positive values suggest a deficit of heterozygotes and the possible presence of inbreeding.
Using the above definition, IC should be a property of the population, and so would be expected to be drawn from the same distribution for all sites and for all variants at a site. Deviations from this distribution can suggest issues in calling a site correctly. Violations of HWE assumptions and/or non-random sampling may adversely affect the distribution of IC, causing it to be shifted. However, outliers can still be identified, although thresholds may need to be adjusted accordingly.
Sites in the output msVCF can be filtered on the following global metrics:
| • | QUAL |
| • | Number of samples with called genotypes (GNS_GT) |
| • | Inbreeding coefficient (GIC) |
| • | X² Hardy-Weinberg Equilibrium P-value (GHWEc2) |
The syntax of a filtering expression is the same as that used by the small variant caller. See Germline Variant Small Hard Filtering. Filters are always applied to the globally-computed metrics, not the values for the current batch. Records failing filter will have the specified filter ID(s) written to the FILTER column of the msVCF, or will be omitted entirely if the --gg-skip-filtered-sites option is specified. Because filtering is on a per-site basis, filters cannot be applied separately to SNPs or indels as they can in the variant caller.
